
Gene Therapy
ICH Q5A revision 2 guidance has recently been updated to include “genetically engineered viral vectors and viral vector-derived products (e.g., viral vaccines, gene therapy products), provided they are amenable to viral clearance, without a negative effect on the product.” Products applicable to revision 2 of ICH Q5A include viral vectors such as adeno-associated virus (AAV) produced using transient or stable transfected cell lines, or by infection using a recombinant virus. Also included are viral vector-derived products such as the following:
- baculovirus-expressed virus-like particles (VLPs),
- protein subunits,
- nanoparticle-based protein vaccines
- and therapeutics.
In addition, the guidance includes AAV gene therapy vectors dependent on helper viruses such as herpes simplex virus or adenovirus for their production.
While the guidance emphasizes the need for gene therapies to demonstrate the robustness of the manufacturing process to remove or inactivate viruses, it does address certain challenges associated with viral clearance. Given gene therapies share similar physiochemical characteristics to viral contaminants, some viral clearance steps have demonstrated limitations regarding their ability to remove or inactivate viruses while maintaining product quality of the gene therapy. When viral clearance is not an option, manufacturers should leverage their risk assessment for mitigation of potential viral contamination by establishing robust viral control strategies that rely heavily on prevention and detection of viral contaminants. All manufacturers of gene therapies should have a comprehensive program for sourcing raw materials, and it is recommended that closed systems be employed during manufacturing. Manufacturers should test process intermediates and critical manufacturing reagents for potential viral contaminants.
When adapting a viral clearance strategy, manufacturers should perform a risk assessment to identify potential sources of viral contaminants in their process. Potential pathways of entry for viral contaminants could include master cell banks and working cell banks that contain endogenous virus like particles produced during the cell culture process. Master virus seeds and working virus seeds could also contain viral contaminants. In addition, raw materials present a pathway for viral contamination. Manufacturers should avoid biologically derived raw materials, if possible, to minimize the potential for viral entry into their processes. Robust testing of cell banks, virus seeds, and raw materials should be implemented to screen for viral impurities. Additionally, viruses can be introduced into a manufacturing process through operators and equipment.
Unique to gene therapies are those platforms that utilize helper viruses in their production to aid in replication and productive infection or those platforms that produce endogenous virus-like particles in the cell culture process. The viral clearance strategy must then demonstrate removal or inactivation of all these classes of viruses identified within the risk assessment by the downstream purification process.
Process robustness in gene therapy manufacturing should be assured by including several orthogonal unit operations in the viral clearance strategy. Unlike mAb processing where viral clearance typically includes dedicated virus inactivation and virus filtration steps, gene therapies often employ a customized viral clearance strategy where the expression system and physicochemical properties of the gene therapy are considered. Inclusion of such steps should be based on product quality by evaluating the physiochemical properties of the gene therapy. For example, if the inclusion of a low pH step for viral clearance substantially decreases the potency of the gene therapy of interest, the step should be omitted from the process. Evaluation of robustness can be demonstrated by the amount of clearance of model viruses or endogenous viruses, such as helper viruses, across different manufacturing unit operations using spiking studies in a validated scaled-down manufacturing process. Challenges associated with viral clearance can arise due to similarities of the test material (gene therapy process intermediates) and viruses. Manufacturers should leverage the physiochemical properties of both the gene therapy and potential viral impurities to ensure clearance of the contaminant without harming the gene therapy.
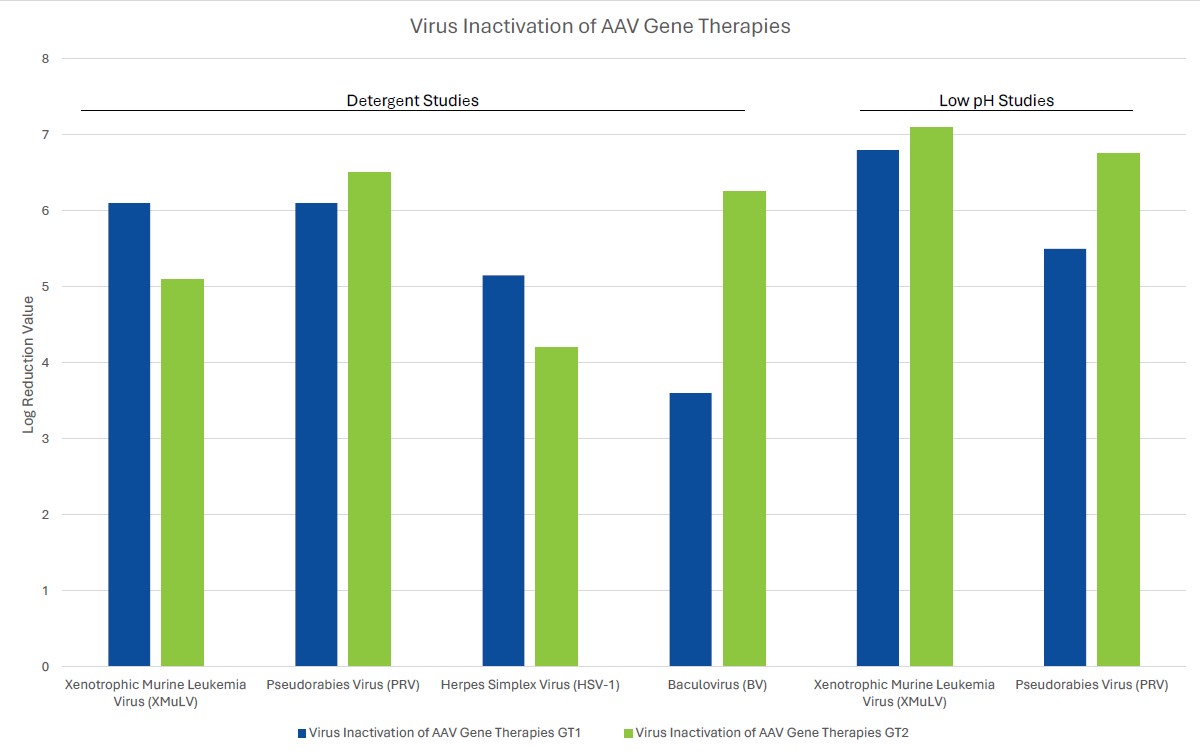
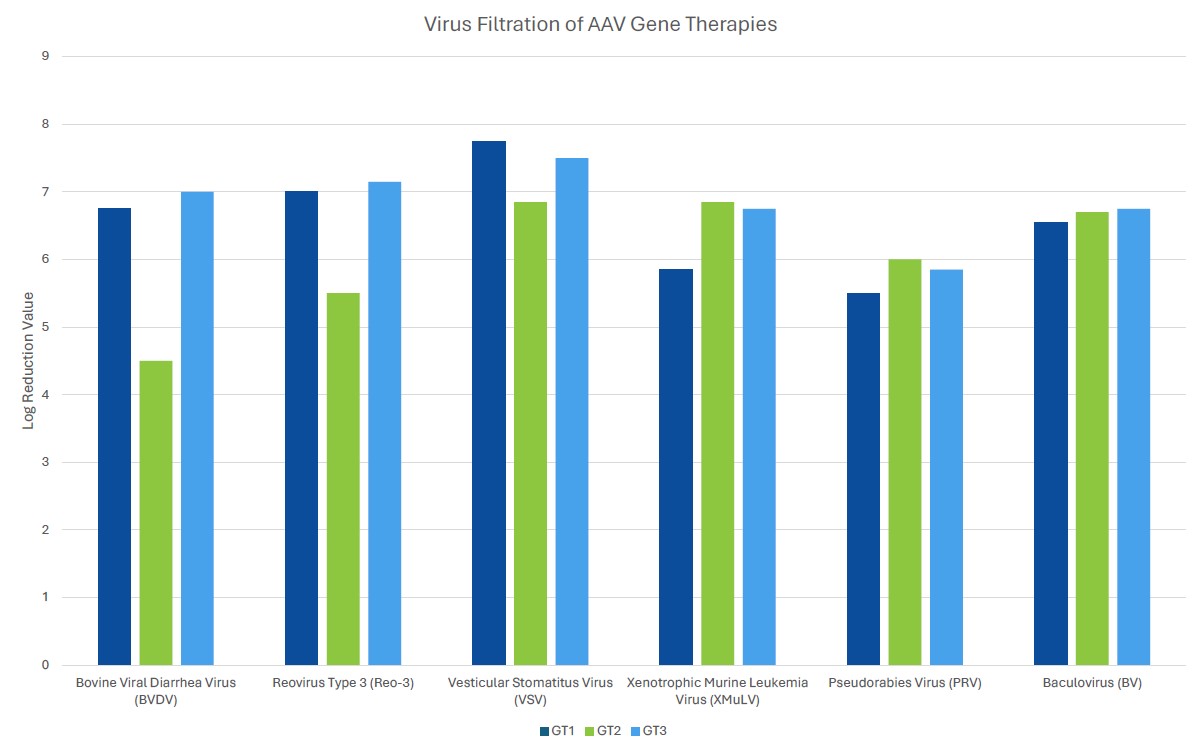
When choosing a virus panel for first-in-human clinical gene therapy viral clearance spiking studies, the risk assessment must be considered, as well as the expression system of the gene therapy. Helper viruses, such as herpes simplex virus or adenovirus, must be included in the virus panel of those AAV gene therapy vectors dependent on these helper viruses to aid in replication. Removal or inactivation of the helper virus by the downstream process must be demonstrated in viral clearance studies. The virus panel of baculovirus-expressed VLPs should include baculovirus and demonstrate its removal or inactivation in the downstream process. Removal or inactivation of insect rhabdovirus which is present in the expression system of Sf9 cells is often evaluated using a model mammalian rhabdovirus. As such, the mammalian rhabdovirus should be included in the panel for those expressions systems utilizing Sf9 cells. The virus panel of other gene therapy expression systems should be considered on a case-by-case basis with the risk assessment supporting selection of spiking viruses. When selecting additional viruses for late-stage viral clearance studies, evaluation of non-specific model viruses with varying physiochemical characteristics should be chosen to demonstrate process robustness with the risk assessment also considered.